
Rett Syndrome: Symptoms and Treatment
What is Rett Syndrome?
Rett syndrome is a genetic disorder characterized by intellectual decline, stereotypical hand movements (such as hand-wringing), slowed head growth, epilepsy, and behavioral and gait disturbances.
Patients with Rett syndrome cannot care for themselves and require constant supervision. With appropriate symptomatic treatment, some people with this condition live for a considerable time.

A child with Rett syndrome. Image edited from a photo: re-cognition.center
The syndrome is named after pediatrician and neurologist Andreas Rett, who observed mentally disabled girls with unusual “hand-washing” type movements [1].
Prevalence of Rett syndrome
The disorder occurs in one out of 10–20 thousand newborns, though this rate is higher in families with consanguineous marriages [2][22].
Rett syndrome causes the majority of cases of intellectual disability among girls [3]. The disease can be masked by autism spectrum disorder (ASD), epilepsy, and psycho-speech developmental delay, making timely and accurate diagnosis difficult.
Thanks to advances in genetic research and diagnostic capabilities, Rett syndrome has become easier to distinguish from other conditions with similar manifestations. This has raised awareness about the condition and led to the creation of organizations such as the International Rett Syndrome Association, the International Rett Syndrome Foundation (IRSF), the Rett Syndrome Research Trust, the Unified Information Resource on MECP2-Related Disorders, the Association for Assistance to Patients with Rett Syndrome, and others [4][5].
Causes of Rett syndrome
In more than 90% of cases, the condition is associated with a spontaneous mutation in the MECP2 gene, which disrupts the synthesis of a protein affecting brain development [3][22]. This gene is always located on the X chromosome. Since the female genome has two such chromosomes, a healthy chromosome partially mitigates the mutation in the second one.
The male genome consists of X and Y chromosomes, so the gene mutation fully affects fetal development, which is why boys more often die in utero [2].
Studying the probability of hereditary transmission of Rett syndrome is difficult because, in the classic course of the disease, people don’t marry due to their severe condition.
However, scientists have identified cases where some maternal cells contain defective DNA.
Therefore, if a family has a child with this diagnosis, genetic testing is advisable when planning a new pregnancy to prevent the birth of another child with this syndrome [8].
Specific factors influencing the occurrence of Rett syndrome have not yet been identified. One can only assume that its development is facilitated by harmful environmental factors, intrauterine infections, and other general risk factors for all congenital genetic diseases [26].
However, it is known that parental age does not affect the development of Rett syndrome [25].
Symptoms of Rett syndrome
If you have similar symptoms, consult a neurologist online: they will provide detailed recommendations. Self-treatment is dangerous.
Rett syndrome is characterized by the following manifestations:
- Loss of acquired skills
- Changes in gait and muscle tone (tension)
- Signs of autism spectrum disorder
- Unusual hand movements
- Epileptic seizures
- Breathing disturbances
- Delayed head growth (microcephaly)
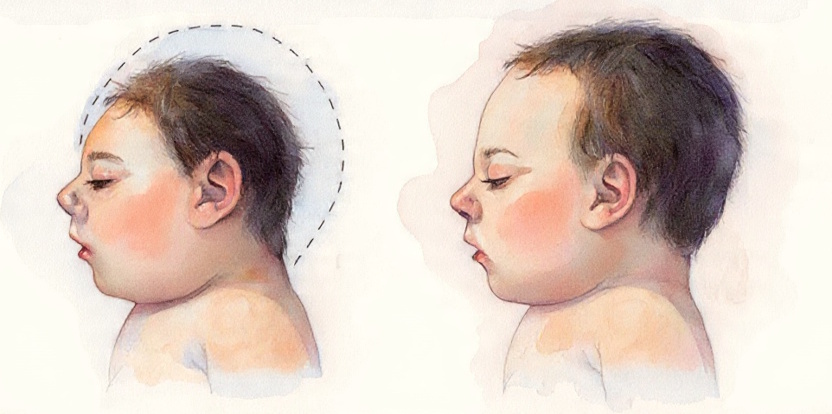
Microcephaly. Image: re-cognition.center
Pregnancy and childbirth typically proceed without complications. For the first 6–20 months, the child doesn’t differ from their peers, but then mental development slows. Head and brain growth lags behind normal rates, and muscle tone decreases [9].
Between ages 1–3 years, developmental regression occurs, and anxiety, sleep disturbances, and frequent unexplained crying may appear.
The child shows signs of autism:
- difficulties in communication,
- avoidance or persistent eye contact,
- absence of pointing gestures and speech.
Facial expressions are scarce with involuntary grimacing.
The child doesn’t respond to their name and doesn’t obey, and their play and interests are monotonous. Purposeful hand movements deteriorate, making it difficult for the child to hold objects and toys.
Stereotypical hand-washing-like movements appear: clenching, clapping, and tapping at body and head level, less commonly finger-sucking and biting.
Over time, the child may stop walking, crawling, and even sitting [10].
Half of children experience breathing disturbances while awake: breathing stops for 1–2 minutes, sometimes leading to fainting, then becomes rapid. Seizures may also occur. These symptoms appear suddenly, within several weeks, leading to incorrect diagnoses, including encephalitis [10].
In elementary school, profound intellectual disability is noted, so children study in special education classes. They exhibit seizures, tremor, disturbances of muscle tone, coordination, and motor skills.
The child walks with widely spread legs and sways, movements become jerky and imprecise. They lose the ability to chew solid food and swallow, and choking occurs. Meanwhile, sleep and emotional contact may improve [10].
By age 10, motor disturbances intensify. Muscle tension increases, joints deform, movement limitations occur (contractures), and scoliosis develops [10].
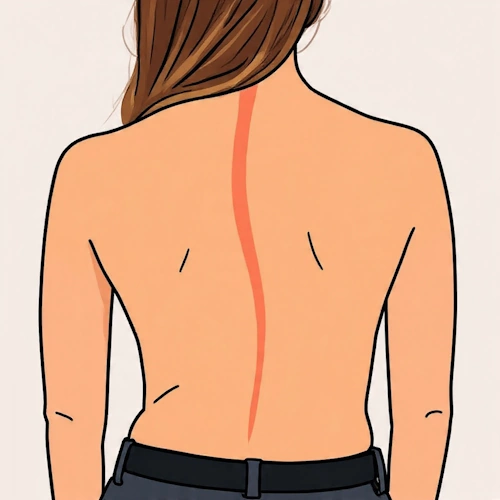
Scoliosis. Image: re-cognition.center
Rett syndrome involves various types of epileptic seizures that respond poorly to anticonvulsant treatment. Most common are generalized tonic-clonic seizures, where abnormal electrical activity is observed in both brain hemispheres. These are accompanied by loss of consciousness, increased muscle tone, and subsequent tremor. Partial seizures without loss of consciousness also occur (in such seizures, epileptic activity arises only in a specific brain area). Sudden falls may happen during epileptic seizures. As the condition progresses, seizures become less frequent.
Sexual development in people with Rett syndrome is normal but is combined with delays in growth and weight gain.
In atypical cases, the condition debuts earlier and manifests immediately after birth. In such severe cases, motor skills may not develop at all: the child doesn’t sit, walk, crawl, or even roll over. Epileptic seizures occur in the first years of life. The child has no period of normal mental development.
Pathogenesis of Rett syndrome
The pathogenesis of the condition is not fully understood. It is based on a random mutation of the MECP2 gene on the X chromosome. This DNA segment is responsible for producing the corresponding protein in many body cells. It is most synthesized in nerve cells (neurons) of the brain, so the protein affects their maturation, plasticity, and formation of connections between them.
In Rett syndrome, the protein structure is disrupted, leading to its malfunction. Brain growth and development suffer, and the cerebral cortex and cerebellum atrophy (shrink) [11]. Thinning of the cerebral cortex leads to disruptions in speech centers and control of behavior and movement planning, while cerebellar atrophy causes coordination problems and tremor.
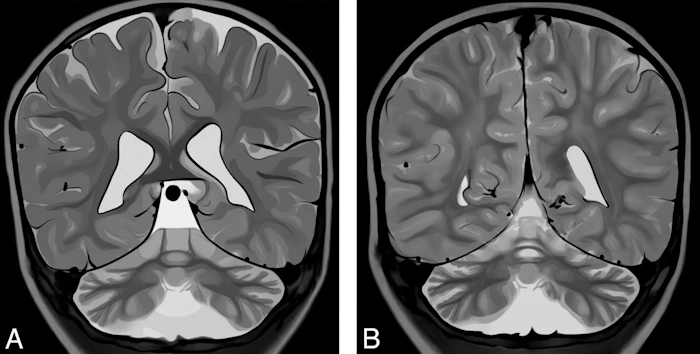
Digital illustration made based on the real image of the brain where atrophy of the cerebellum and cerebral cortex is shown. Image: re-cognition.center
Classification and stages of Rett syndrome development
Rett syndrome has several stages:
- Stage I — “stagnation.” This lasts several months after disease onset and manifests as delayed mental development and autism spectrum disorder-type behavioral disturbances. Slowed head growth and decreased muscle tone are observed.
- Stage II — “rapid regression.” This stage lasts from several weeks to several months, rarely years. Autistic features soften, the child shows emotions in close surroundings, but social contact is impaired. Speech becomes slurred, motor clumsiness and general excitability appear. Hand movements are uncontrolled, leg muscles atrophy, gait is disturbed, and scoliosis develops. The child shows first signs of breathing disturbances, and microcephaly forms.
- Stage III — “pseudostationary.” The third stage lasts over 10 years and is characterized by complete absence of speech and significant intellectual decline. The child can understand simple gestures, moves hands obsessively less, and develops tremor of the head, body, and limbs. One-third of patients begin having epileptic seizures [10]. At this stage, developmental regression occurs to the level of a one-year-old child.
- Stage IV — “total dementia.” During this period, most necessary daily living skills, swallowing, and chewing disappear. Limb tone significantly increases. Due to lack of motor activity, joint contractures, weakness, and muscle hypertonia, the child needs constant care. Even with this, the patient may die between ages 10–30 [10].
These stages are distinguished conditionally because the boundaries are unclear and age ranges may shift.
Besides the classic course, atypical forms of Rett syndrome are identified:
- Zapella — with partially preserved speech. Dementia is less pronounced, physical development doesn’t suffer.
- Hanefeld — with early onset of seizures, sometimes even before intellectual disability appears.
- Rolando — with predominant motor development disturbances [6].
Atypical forms of Rett syndrome also include cases of early manifestation onset (from birth or in utero) and its detection in live-born boys, since many boys with this syndrome die in utero [2].
All atypical forms meet the criteria for Rett syndrome, but the disease dynamics deviate from the classic course. This complicates differential diagnosis with autism spectrum disorder and hereditary epilepsy.
Complications of Rett syndrome
Complications may appear from Stage II of the disease and affect nearly all organs. The patient’s condition is worsened by:
Breathing and swallowing disorders. Breathing problems are associated with impaired nervous regulation and scoliosis. They can lead to fainting and sudden death. Swallowing disturbances cause pneumonia, and air swallowing causes abdominal bloating. Additionally, sleep disorders accompanied by breathing abnormalities worsen epilepsy course and intellectual decline [13].
- Scoliosis. It occurs due to changes in tone and muscle atrophy. With severe spinal curvature, chronic pain appears and breathing disturbances worsen. To ease the condition and slow progression, braces are used and surgeries are performed [14].
- Joint contractures. Due to increased muscle tone, the patient must exert more effort to move joints. As the disease progresses, the joint deforms so much that movements are limited or cannot be performed at all. To prevent contractures, massage, physical therapy, Botulinum toxin injections, orthoses, and other orthopedic devices are used. If a contracture has already formed, in some cases it is surgically corrected [15].
- Bone fractures. Leg bones break most often because bone mineral density is very low. Patients with Rett syndrome experience significantly delayed skeletal maturation and bone mass accumulation. This occurs due to anticonvulsant medication use, joint contractures, low physical activity, poor nutrition, and low calcium and vitamin D intake [16].
- Status epilepticus. In this condition, a prolonged epileptic seizure or continuous series of seizures occurs, resulting in brain neuron damage. This complication can significantly worsen the condition or even lead to death [17].
- Impaired blood circulation in arms and legs. Vascular tone regulation problems lead to spasm. Growth of hands and feet slows, they become small, cold, and cyanotic [2].
- Muscle atrophy. Muscle mass gradually decreases due to low physical activity, impaired nervous regulation, and nutrient deficiency [2].
- Cachexia. Body mass deficit is associated with behavioral disturbances, selective eating, and loss of ability to chew and swallow.
- Constipation and abdominal pain. Gastrointestinal (GI) tract disruptions occur due to eating problems, decreased motor activity, and changes in intestinal microflora [18].
- Pressure sores. They form in bedridden patients. For prevention, position must be changed frequently, bedding condition monitored, and skin defects treated.
Diagnosis of Rett syndrome
Parents typically consult a pediatrician or pediatric neurologist with complaints about developmental delays. When signs of autism (communication disturbances) and severe mental developmental delays are identified, the child may be referred to a psychiatrist. If doctors suspect Rett syndrome, the patient’s condition is compared against special criteria to confirm or rule out the diagnosis.
Diagnostic criteria for Rett syndrome
Main criteria:
- Partial or total deterioration of previously learned, purposeful hand abilities.
- Speech problems or absence
- Gait disturbances up to inability to walk
- Stereotypical hand movements: rubbing, clenching, clapping, tapping, and licking [12]
Additional criteria:
- Breathing disturbances while awake
- Bruxism (teeth grinding) while awake
- Sleep problems
- Changes in muscle tone
- Vascular disturbances causing blueness of hands, feet, or other body parts
- Scoliosis and/or kyphosis (spinal curvature toward the back)
- Growth delay
- Small cold hands and feet
- Inappropriate bursts of laughter/screaming
- Reduced pain response
- Very persistent eye contact or its absence
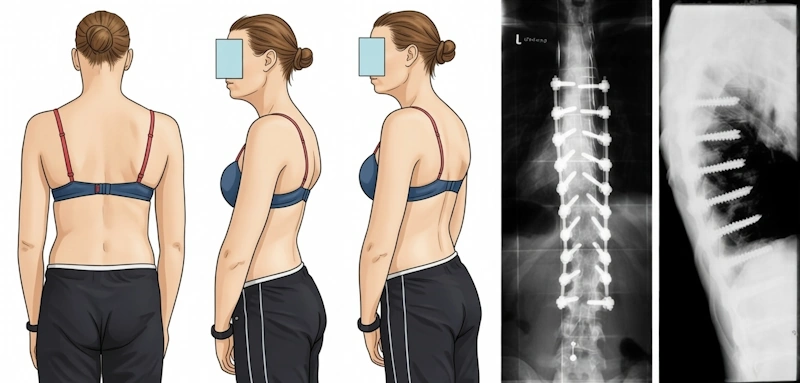
Kyphosis. Image: re-cognition.center
Criteria excluding Rett syndrome:
Brain damage resulting from trauma (before or after birth), severe infection, or neurometabolic disease (nervous system damage due to metabolic disorders)
For diagnosing typical Rett syndrome, the following conditions must be met:
- Period of regression followed by recovery or stabilization
- Presence of all mandatory criteria and absence of excluding criteria
- Possible presence of additional criteria (not required)
To diagnose an atypical form of Rett syndrome, the following must be observed:
- Period of regression followed by recovery or stabilization
- At least 2 of 4 mandatory criteria and 5 of 11 additional criteria
- Absence of excluding criteria
Additional examinations
When Rett syndrome is suspected, genetic testing is performed for MECP2 gene mutation on the X chromosome (Xq28) and for other presumed genetic mutations.
Lung function and breathing disturbances are investigated using spirometry.

How is spirometry done. Image: re-cognition.center
When performing an electroencephalogram (EEG), most patients show decreased baseline activity in the background recording, indicating slowed neuronal activity. Some people with Rett syndrome show epileptiform activity characteristic of epilepsy, but this doesn’t always confirm its presence. Video-EEG monitoring shows the connection between seizures and EEG changes [10].
To identify complications or for differential diagnosis, blood tests, electrocardiography (ECG), ultrasound examination of abdominal organs, bone X-rays, and magnetic resonance imaging (MRI) are performed.
Differential diagnosis
Differential examination is conducted with autism spectrum disorder [19]. In autism, there is no pronounced negative dynamics, but there is developmental regression in the first years of life. ASD can be accompanied by epilepsy, but in such cases it responds better to treatment.
Autism spectrum disorder is more common in boys, so when diagnosing girls with signs of autism, more attention should be paid to exclude Rett syndrome. Children with this disease experience deterioration of motor skills and ataxia after the autistic regression stage. Then seizures occur, which are not characteristic of ASD.
The combination of epilepsy, profound intellectual disability, gait disturbance, and automatic hand movements in autism requires thorough investigation to consider a diagnosis of Rett syndrome.
Epilepsy poorly responsive to therapy, combined with developmental delays, also requires exclusion of Rett syndrome.
Treatment of Rett syndrome
Do not self-medicate
Consult an experienced doctor. This can be done online.
Treatment that eliminates the underlying cause of the disease does not yet exist. Scientists are studying the effectiveness and safety of gene therapy for Rett syndrome, but it is not yet used in clinical practice [20]. Patients receive symptomatic treatment to slow disease progression and its transition to a severe stage, eliminate seizures, and maintain internal organ function.
Symptomatic therapy includes medication, physical therapy, and correction of mental development deviations.
The following medications are used to soften Rett syndrome manifestations:
- Anticonvulsants — reduce seizure frequency
- Proton pump inhibitors and H2-blockers — used for gastric content reflux into the esophagus
- Laxatives — help with constipation
- Melatonin — improves sleep quality
- SSRI antidepressants (selective serotonin reuptake inhibitors) — prescribed for increased anxiety [21]
- Risperidone neuroleptic — stops self-injury from uncontrolled hand movements and overexcitation
- Atropine — helps with excessive salivation
- Multivitamins and vitamin D — strengthen bones and replenish deficiencies
- Medications correcting breathing and heart problems. Oxygen therapy and mechanical ventilation are also used for these disturbances
In the USA, the first medication for treating Rett syndrome has been approved — Trofinetide. Its clinical trial is still ongoing. The drug is expected to improve cerebral cortex plasticity and nerve impulse transmission [21].
As part of physical therapy for Rett syndrome, therapeutic exercises and massage are performed to slow the progression of motor disturbances, reduce muscle tone, and prevent contractures and bone fragility.
Psychologists and special education teachers work with patients to reduce the rate of intellectual disability development [2]. These rehabilitation measures include developing attention, memory, thinking, social adaptation, and teaching speech, writing, counting, and daily living skills.
Prognosis. Prevention
The prognosis primarily depends on the form of the disease and complications that arise, but quality of care also matters. Children with Rett syndrome sometimes live to 20–30 years and die from cachexia, respiratory disorders, and during status epilepticus. Bedridden patients may die during flu or other acute respiratory viral infections due to aspiration pneumonia.
Some people with this disease can move independently, others only in a wheelchair [2].
In mild forms of the disease, minimal positive developmental results can be achieved.
Gene therapy research may offer hope for extending and improving quality of life for people with Rett syndrome.
There are no specific prevention options, but in families with a child with Rett syndrome, mothers undergo prenatal diagnosis to make decisions about continuing or terminating pregnancy [8].
Sources
1. Rett A. Uber ein eigenartiges hirnatrophisches Syndrom bei Hyperammonämie im Kindersalter [On a unusual brain atrophy syndrome in hyperammonemia in childhood] // Wien Med Wochenschr. — 1966. — № 37. — Р. 723–726.
2. Kazantseva L. Z., Ulas V. Yu. Rett syndrome in children // Attending Physician. — 1998. — № 6. — P. 12–16.
3. Liyanage V. R., Rastegar M. Rett syndrome and MeCP2 // Neuromolecular Med. — 2014. — № 2. — Р. 231–264.
4. Hunter K. Role of the International Rett Syndrome Association // J Child Neurol. — 1988. — № 3. — Р. 87–88.
5. Downs J., Pichard D. C., Kaufmann W. E., Horrigan J. P. et al. International workshop: what is needed to ensure outcome measures for Rett syndrome are fit-for-purpose for clinical trials? June 7, 2023, Nashville, USA // Trials. — 2024. — № 1. — Р. 845.
6. Volgina S. Ya. Atypical variants of Rett syndrome // Rett Syndrome. [Electronic resource]. Access date: 17.07.2025.
7. And now for something new (bed sores) // Grace for Rett Syndrome. — 2016.
8. Armstrong J., Aibar E., Pineda M., Pérez M. M. et al. Prenatal diagnosis in Rett syndrome // Fetal Diagn Ther. — 2002. — № 4. — Р. 200–204.
9. Shnayder N. A. Rett syndrome (part 1): definition, epidemiology, etiology, clinic // Bulletin of KB № 51. — 2014. — № 1. — P. 18–25.
10. Mukhin K. Yu., Karpova V. I., Bezrukova I. S., Bolshakova E. S. et al. Rett syndrome (literature review and clinical case description) // Russian Journal of Pediatric Neurology. — 2010. — № 2. — P. 43–52.
11. Kyle S. M., Vashi N., Justice M. J. Rett syndrome: a neurological disorder with metabolic components // Open Biol. — 2018. — № 2. — Р. 170216.
12. Neul J. L., Kaufmann W. E., Glaze D. G., Christodoulou J. et al. Rett syndrome: revised diagnostic criteria and nomenclature // Ann Neurol. — 2010. — № 6. — Р. 944–950.
13. Ramirez J. M., Karlen-Amarante M., Wang J. J., Huff A. et al. Breathing disturbances in Rett syndrome // Handb Clin Neurol. — 2022. — Vol. 189. — Р. 139–151.
14. Huang T. J., Lubicky J. P., Hammerberg K. W. Scoliosis in Rett syndrome // Orthop Rev. — 1994. — № 12. — Р. 931–937.
15. Guidera K. J., Borrelli J. Jr., Raney E., Thompson-Rangel T. et al. Orthopaedic manifestations of Rett syndrome // J Pediatr Orthop. — 1991. — № 2. — Р. 204–208.
16. Caffarelli C., Al Refaie A., Mondillo C., De Vita M. et al. Bone Fracture in Rett Syndrome: Mechanisms and Prevention Strategies // Children (Basel). — 2023. — № 12. — Р. 1861.
17. All-Russian Society of Neurologists, Association of Neurosurgeons of Russia, Association of Clinical Neurophysiology Specialists, Union of Rehabilitologists of Russia. Epilepsy and status epilepticus in adults and children: clinical guidelines. — 2022. — 277 p.
18. Caputi V., Hill L., Figueiredo M., Popov J. et al. Functional contribution of the intestinal microbiome in autism spectrum disorder, attention deficit hyperactivity disorder, and Rett syndrome: a systematic review of pediatric and adult studies // Front Neurosci. — 2024. — № 18. — Р. 1341656.
19. Trevathan E., Naidu S. The clinical recognition and differential diagnosis of Rett syndrome // J Child Neurol. — 1988. — № 3. — Р. 6–16.
20. Powers S., Likhite S., Gadalla K. K., Miranda C. J. et al. Novel MECP2 gene therapy is effective in a multicenter study using two mouse models of Rett syndrome and is safe in non-human primates // Mol Ther. — 2023. — № 9. — Р. 2767–2782.
21. Percy A. K., Ananth A., Neul J. L. Rett Syndrome: The Emerging Landscape of Treatment Strategies // CNS Drugs. — 2024. — № 11. — Р. 851–867.
22. Operto F. F., Mazza R., Pastorino G. M. G., Verrotti A. et al. Epilepsy and genetic in Rett syndrome: A review // Brain Behav. — 2019. — № 5. — Р. e01250.
23. About Rett Syndrome // IRSF. [Electronic resource]. Access date: 17.07.2025.
24. Vedolin L., Gonzalez G., Souza C. F., Lourenço C. et al. Inherited cerebellar ataxia in childhood: a pattern-recognition approach using brain MRI // AJNR Am J Neuroradiol. — 2013. — № 5. — Р. 925–934.
25. Asuncion R. M. D., Ramani P. K. Rett Syndrome // Treasure Island (FL). — 2025.
26. Congenital disorders // World Health Organization.
Information presented on the site cannot be used for diagnosis, treatment prescription, and does not replace a doctor’s visit.